Beim Braunvieh existieren zwei seltene Farbvarianten: Blüem und Gurt. In diesem Blogbeitrag beschreiben wir die indirekte Ableitung des Blüem-Genotyps. Blüem-Tiere sind gekennzeichnet durch eine reduzierte Pigmentierung. Wie in Abbildung 1 gezeigt, sind Haare im Bereich des Kopfs, der Beine und der Wirbelsäule nicht pigmentiert und erscheinen daher weiss.

Blüem wird dominant vererbt. Deshalb zeigen auch heterozygote Tiere die charakteristische Färbung. Die dominante Vererbung machte es den Züchtern einfach, die Blüem-Farbvariante in der Population aufrechtzuerhalten. Andererseits verhindert sie die Unterscheidung zwischen reinerbigen und mischerbigen Tieren anhand der Fellfarbe.
Mit der indirekten Bestimmung des Genotyps wollen wir diese Lücke schließen und den Züchtern die Differenzierung zwischen homo- und heterozygoten Tieren ermöglichen. Homozygote Blüem-Tiere sind für die Züchter deswegen von besonderem Interesse, da sie unabhängig vom Besamungsstier ausschließlich Nachkommen zur Welt bringen, welche die Blüem Färbung im Haarkleid ausprägen.

Abbildung 1: Pigmentierung bei einem Blüem-Tier.
Blickt man ins Detail, ist die Genetik hinter der Blüem-Färbung sehr speziell. Ursache ist eine komplexe strukturelle Variante (Cs6). Dabei wurde ein mehrere Tausend Basenpaar langes Segment vom Chromosom 6 umarangiert auf Chromosom 29 integriert, bevor es wieder zum Chromosom 6 zurückgebaut wurde (Durkin et al 2012, Abb. 2). Die Färbung des Haarkleids ist deswegen betroffen, weil in dem Segment das Farbgen KIT enthalten ist. Als Konsequenz daraus haben Blüem-Tiere somit ein Abschnitt im Erbgut mehrfach.
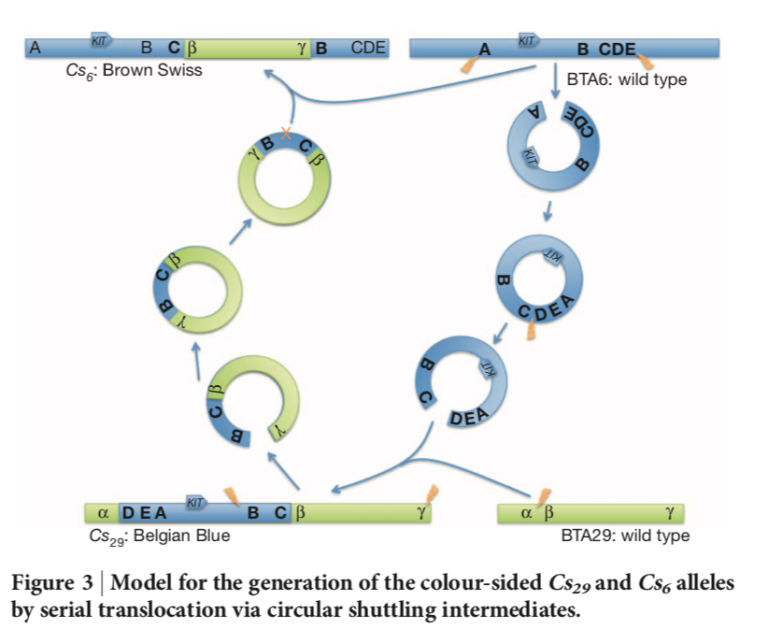
Abbildung 2: Durkin et al 2012.
Aus dem Datensatz der genomischen Selektion stehen von mehreren Tausend Tieren hoch-dichte SNP-Genotypen zur Verfügung. Dabei werden in der Regel Illumina® Chips zur Typisierung verwendet. Bei der Typisierung mit Illumina®-Chips erfolgt die eigentliche Bestimmung jedes einzelnen Genotyps mittels Farbsignalen bzw. Clusteranalysen auf Basis von Signalintensitäten. So stehen aus der Genotypisierung neben den eigentlichen Genotypen zusätzlich normalisierte Farbintensitäten (R) und Intensitätsquotienten (θ) zur Verfügung. Diese Werte werden stets in Relation zu einer Standardreferenz ausgegeben.
Strukturelle Varianten, welche eine Mindestgröße überschreiten, können somit anhand der log(R)-Ratios in den Illumina®-Chip Ergebnissen erkannt werden. SNP in multiplizierten Regionen (z. B. durch strukturelle Variation) haben erhöhte Signalintensitäten im Vergleich zum Referenz-Panel. Die Blüem-Translokation ist gross genug und kann so in den Signalintensitäten, wie in Abbildung 3 gezeigt, leicht erkannt werden.
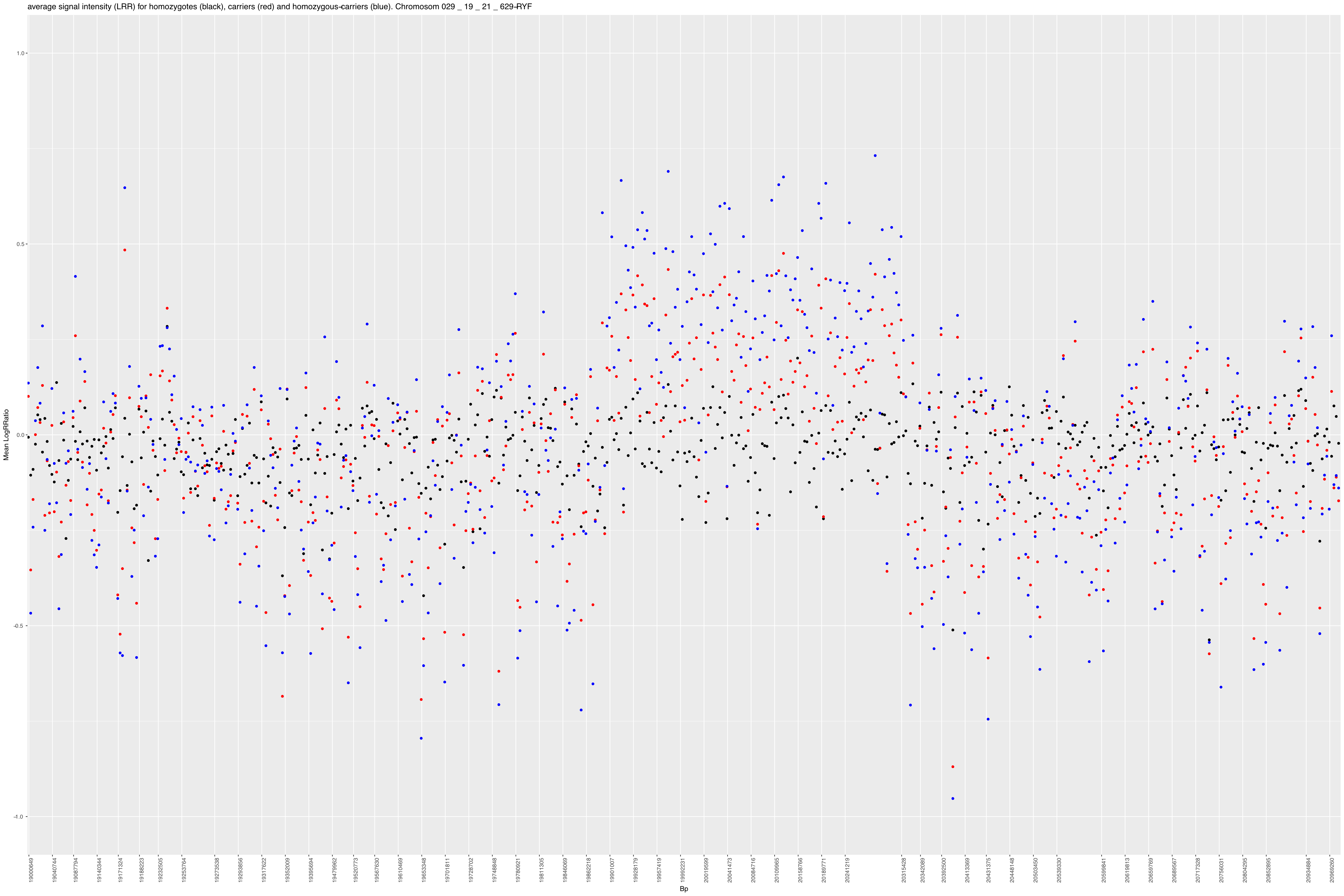
Abbildung 3: Durchschnittliche Signalintensitäten pro SNP in Abhängigkeit des CS6 – Genotyps (schwarz: wt/wt, rot: wt/CS6, blau: CS6/Cs6).
Wir benutzen Support Vector Maschine (SVM) Algorithmen zur indirekten Ableitung des Blüem-Genotyps. SVM Algorithmen gehören in den Bereich des Supervised Learning, wobei immer auf der Basis von Referenzdaten eine vorhersagende Funktion abgeleitet wird. Für jede Beobachtung wird eine Response (in unserem Beispiel dem Blüem-Genotyp) erwartet. Oftmals wird der Referenzdatensatz auch Trainingsdatensatz genannt, denn diese Beobachtungen werden verwendet, um mit der jeweiligen Methode die Funktion ƒ herzuleiten. Die vorhersagende Funktion ƒ akzeptiert als Input die Eigenschaften der Beobachtungen, welche auch als Features bezeichnet werden. In unserem Beispiel entsprechen die Features den log(R)-Ratios aus der Typisierung mit einem Illumina® Chip. Als Output liefert die Funktion ƒ die Zugehörigkeit der Beobachtung zu einer bestimmten Gruppe. Für unser Beispiel entsprechen die Gruppen den verschiedenen Blüem-Genotypen.
Die verschiedenen Methoden aus dem Bereich des Statistical Learnings unterscheiden sich in der Vorhersagbarkeit und der Interpretierbarkeit. SVM Algorithmen, insbesondere unter Verwendung von nicht linearen Kernel, sind charakterisiert durch eine hohe Flexibilität wobei die Funktion an sich eher schwer zu interpretieren ist.
In unserem Beispiel setzt sich das Trainingsset aus Tieren zusammen, die sowohl Illumina-Chip Daten als auch Cs6 Genotypen haben. Die Zusammenstellung ist in Tabelle 1 abgebildet.
Trainingsset mit Illumina-Chip Daten und Cs6 Genotypen
|
Phänotyp |
Cs6 – Genotyp |
Anzahl Referenz Tiere |
| Braun | wt/wt | 2003 |
| Blüem | wt/Cs | 87 |
| Blüem | Cs/Cs | 19 |
Tabelle 1: Zusammensetzung Referenzdatensatz.
Die Signalintensitäten der SNPs im translozierten Abschnitt werden als Beobachtung behandelt. Der Cs6-Genotyp ist die Response.
Damit haben wir in unserem Fall eine kategorische Grösse als Response und damit einen Klassifizierungsansatz. Ausserdem ermöglicht der SVM Ansatz eine Klassifizierung unter Verwendung von nicht linearen Zusammenhängen. Evaluiert wurde der Ansatz einerseits unter Verwendung verschiedener Chip-Dichten (Illumina HD, 150K, bzw. LD-SNP Dichte), sowie andererseits unter Anwendung zweier unterschiedlicher Kernel-Funktionen linear bzw. radial.
Die Methode wurde validiert mit dem Leave-one-out Ansatz. Dafür wurde jedes Tier einmal vom Training ausgeschlossen. Im Folgenden wurde dessen Blüem-Genotyp hergeleitet. Dabei wurde die vorhersagende Funktion ƒ des SVM-Algorithmus angewendet, welche an Hand der verbliebenen n-1 Traininsgtiere abgeleitet wurde. Anschliessend wurde der hergeleitete Genotyp mit dem originalen Genotyp verglichen. Ein leichter Einfluss der Chip-Dichte war erkennbar.
Wie erwartet zeigte der Chip mit der niedrigsten Anzahl SNP die höchste Fehlerquote. Ebenfalls erreichte der lineare Kernel die grösste Genauigkeit. Anhand der Zusammenhänge, wie sie in Abbildung 2 dargestellt sind, entsprach dies den Erwartungen. Alles in allem aber war die Genauigkeit über alle drei Chip-Dichten sehr hoch. Dies geht aus Tabelle 2 hervor. Illumina® Daten scheinen daher zusammen mit SVM-Algorithmen geeignet zu sein, um komplexere strukturelle Varianten ab einer gewissen Grösse herzuleiten.
Anteil (%) korrekt hergeleitete Cs6-Genotypen aus dem Leave-One-Out Ansatz
|
Chip |
Cs6-Genotype prediction |
|
|
SVM Kernel |
Linear |
Radial |
|
777K |
99.8 |
99.7 |
|
150K |
99.8 |
99.8 |
|
LD |
95.8 |
97.2 |
Tabelle 2: Zusammenfassung der Ergebnisse aus der Validierung.

Die Prozedur haben wir in den Ablauf der genomischen Selektion implementiert. Jedes neu typisierte Tier aus dem Routineprozess der genomischen Selektion wird analysiert. Die SVM-Prediction an sich wird im open source framework R(v3.1.0) und unter Verwendung des packages „e1071“ durchgeführt. Die Laufzeit auf einem Linux-Server mit 528 Gb RAM beträgt ohne Parallelisierung zwei Minuten für derzeit rund 51’000 typisierte Tieren, ausgehend von 2’000 Trainingstieren.
Referenz:
- Durkin et al. 2012: Nature. 2012 Feb 1;482(7383):81-4: Serial translocation by means of circular intermediates underlies colour sidedness in cattle
- Illumina Tecnote: https://www.illumina.com/Documents/products/technotes/technote_cytoanalysis.pdf
- R: https://www.r-project.org/
- R-package „e1071“: https://cran.r-project.org/web/packages/e1071/index.html

